

Ligand- Residue Interaction
Mapping Analogous Heterogens onto Residue Interaction

Expertise
Especially in structural bioinformatics and fragment based drug discovery


|
Mapping Analogous Heterogens onto Residue Interactions (Mahori)
Summary: A web service for identification of ligand &
residue interactions. It offers the link between pharmaceutically important
fragments to all biomolecular binding partners in the PDB.
“
Crystal
structures serve as a template for many facets of drug discovery (Breitenlechner et al., 2005). A survey on market value of small-molecule
drugs has shown that two-thirds of the sales resulted from analogue design (Wermuth, 2006). Understanding of i nteractions
made by chemical analogues presented in the Protein Data Bank may suggest
synthetic strategies for lead optimisation. Nevertheless, gathering binding
characteristics of a particular analogue is time-consuming and there is no publicly
available resource that facilitates this type of understanding. Our web-based
tool, Mahori, is acronymed from its function for Mapping Analogous Heterogens
onto Residue Interactions. It aims to provide visualisation and classification
of molecular interactions made by the user-query atoms obtained from the
heterogen section (HET) of the PDB file (wwPDB, 2007). There exists a few web resources that allow
for querying PDB structures and their superpositions based on the ligand
structure; these include Relibase (Bergner et al., 2001) and IsoStar (Bruno et al., 1997). Selection of amino acid residues that
interact with a queried ligand can also be achieved by FireDB (Lopez et al., 2007) and classification of ligand-protein
interactions based on the residue contact can be obtained from MSDsite (Golovin et al., 2005). However, these websites do not allow for
comparing molecular interactions of multiple structures at the level of ligand substructure.
The rationale to allow for user-defined
substructure comparison is based on the idea of pharmacophore, which shares a similar
number and position of interactions such as hydrogen bonds. Selection of the
analogous part of the molecule could give an indication of the significant
interactions that the substructure can contribute to the binding. Analysing all
the contacts made by the whole ligand is also available from our website.
However, the user might be overwhelmed by the amount of weak residue
interactions that may not greatly contribute to the ligand binding. Therefore,
selecting either atoms that comprise the same analogue across several
structures or non-carbon atoms that have higher tendency to make significant
interactions is recommended. Mahori supports a robust querying against
the whole PDB by its underlying PDB-ligand database called Credo developed by
Adrian Schreyer. This database stores all the interactions the ligand make
using the method adapted from interaction types assignment described in the
approach for optimising fragment and scaffold docking (Marcou and Rognan, 2007). Every
atom of the heterogen and its contacting neighbour atoms have their pre-defined
types and the distances are calculated for every interacting pair. By
prioritising the types, the distance and the angle in the case of Pi-Pi
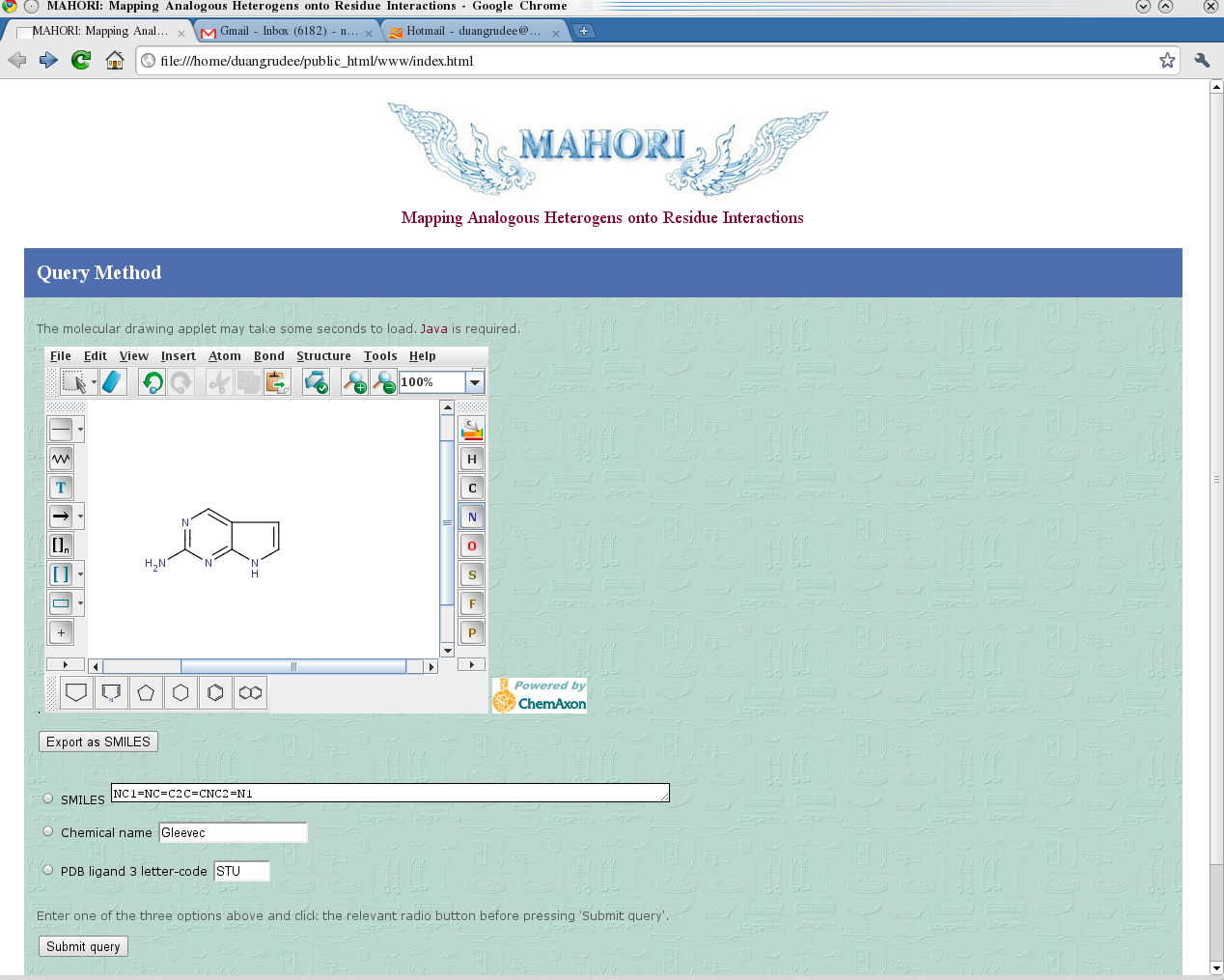
interaction, the interaction types can be assigned for all atom pairs. The user can make a query by providing a chemical
structure, a SMILES string, or a PDB ligand three-letter code, which will bring
up a substructure selection panel. Interacting residues and the type of
interactions made by the user-defined substructure can be retrieved and displayed
in the same page. An example to illustrate the interaction
interpretation is the general kinase inhibitor Staurosporine, which can be
queried from Mahori using the three-letter code STU. The number of hydrogen
bonds that residues around N4 of Staurosporine made corresponds well with the
trend of binding affinitites. Kinase structures that have two residues making hydrogen
bonds to N4 of staurosporine (i.e. PDB code 1AQ1, 1NVR, 1STC, 1OKY, 1YHS), or make
hydrogen bonds with both N4 and O6 (i.e. PDB code 1SM2, 1QPD), all have binding
affinities below 50 nM. The majority (6/7) of structures have only one residue
contribute in the hydrogen bonds to N4 have binding affinities between than
50-820 nM (i.e. PDB code 1BYG, 2DQ7, 2CLQ, 1NXK, 1U59, 2BUJ). Structures that
do not make any hydrogen bond with N4 at all have binding affinities 9000 nM
(PDB code 1E8Z). Therefore, if one wishes to modify Staurosporine to achieve
better affinity for kinase in the latter group, the best strategy is to
identify a residue around the N4 region which can make another hydrogen bond
with the modified staurosporine structure. The comparisons of the amount, the type,
and the position of interactions can be achieved from one resulting page on the
computer screen. This kind of information might be useful for rational drug
design and medicinal chemistry education. References: Bergner, A., Gunther, J., Hendlich, M., Klebe, G. and Verdonk, M. (2001)
Use of Relibase for retrieving complex three-dimensional interaction patterns
including crystallographic packing effects. Biopolymers,
61, 99-110. Breitenlechner, C.B., Bossemeyer, D. and
Engh, R.A. (2005) Crystallography for protein kinase drug design: PKA and SRC
case studies. Biochimica et Biophysica
Acta (BBA) - Proteins & Proteomics Inhibitors of Protein Kinases (4th International Conference, Inhibitors
of Protein Kinases) and Associated Workshop: Modelling of Specific Molecular
Recognition Processes (Warsaw, Poland, June 25-29, 2005), 1754,
38-49. Bruno, I.J., Cole, J.C., Lommerse, J.P.M.,
Rowland, R.S., Taylor, R. and Verdonk, M.L. (1997) IsoStar: A library of
information about nonbonded interactions. Journal
of Computer-Aided Molecular Design, 11,
525-537. Golovin, A., Dimitropoulos, D., Oldfield,
T., Rachedi, A. and Henrick, K. (2005) MSDsite: a database search and retrieval
system for the analysis and viewing of bound ligands and active sites. Proteins, 58, 190-199. Lopez, G., Valencia, A. and Tress, M.
(2007) FireDB--a database of functionally important residues from proteins of
known structure 10.1093/nar/gkl897. Nucl. Acids Res., 35,
D219-223. Marcou, G. and Rognan, D. (2007) Optimizing
Fragment and Scaffold Docking by Use of Molecular Interaction Fingerprints. J. Chem. Inf. Model., 47, 195-207. Wermuth, C.G. (2006) Similarity in drugs:
reflections on analogue design. Drug
Discovery Today, 11, 348-354. wwPDB. (2007) Heterogen Section. Protein Data Bank Contents Guide: Atomic Coordinate Entry Format Description Version 3.0.1, Vol. 2007.
|
|
If you want to cite MAHORI:
Tanramluk D, Schreyer A, Pitt WR, Blundell TL On the origins of enzyme inhibitor selectivity and promiscuity: a case study of protein kinase binding to staurosporine.
Chemical Biology & Drug Design 2009 Jul; 74(1):16-24. Mahori Project
PDB Ligand Update: 10/07/2014 By Duangrudee Tanramluk,Lalita Narupiyakul, Ruj Akavipat, Sungsam Gong Thanks to Tom Blundell at University of Cambridge MB a nd ICBS at Mahidol University
|